Actu CAP - avril 2025

Sommaire
Que retenir des nouvelles recommandations pour le diagnostic et la prise en charge de l’XLH ?
Très attendues, les nouvelles recommandations internationales pour le diagnostic et la prise en charge de l’hypophosphatémie liée à l’X (XLH) actualisent les directives de 2019. Conçues pour optimiser la prise en charge des patients, elles mettent en avant les nouvelles thérapeutiques et la transition enfant-adulte. Élaborées à partir d’un processus structuré initié en 2022 – comprenant une série de conférences en ligne réunissant experts et associations de patients –, ces recommandations ont obtenu la validation de plusieurs sociétés savantes européennes et internationales.
L’XLH est une maladie rare causée par des variants pathogènes du gène PHEX (phosphate-regulating endopeptidase homologue X-linked), principalement exprimé dans l’os (ostéoblastes et ostéocytes) et les dents (odontoblastes et cémentoblastes). L’incidence de l’XLH est estimée à 3,9 pour 100 000 naissances vivantes, et sa prévalence varie entre 1,7 et 4,8 pour 100 000 personnes.
Un mécanisme physiopathologique complexe
L’XLH est principalement caractérisée par une augmentation de la sécrétion du facteur de croissance fibroblastique 23 (FGF23) par l’os, qui, en se liant au récepteur FGF1 et au co-récepteur α-Klotho, entraîne :
- Une réduction de l’expression des transporteurs rénaux du phosphate NPT2a (SLC34A1) et NPT2c (SLC34A3), conduisant à une fuite rénale de phosphate.
- Une réduction de la synthèse de la 1,25-dihydroxyvitamine D (1,25(OH)₂D) accompagnée d’une augmentation de sa dégradation, aboutissant à une hypovitaminose D.
Ce déséquilibre provoque une hypophosphatémie chronique, entraînant des conséquences cliniques majeures dès la petite enfance telles que :
- Rachitisme, ostéomalacie et douleurs osseuses.
- Déformations des membres inférieurs (genu varum/valgum).
- Fusion prématurée des sutures crâniennes et nanisme disproportionné dès les deux premières années de vie.
- Hypominéralisation dentaire, avec un risque accru d’abcès dentaires et de parodontites.
- À l’âge adulte : pseudofractures, ostéoarthrite, enthésopathies, sténose rachidienne, perte auditive, troubles dépressifs et altération de la qualité de vie.
Des défis persistants dans le diagnostic de l’XLH
Le retard de diagnostic reste une problématique majeure. L’XLH est souvent confondue avec d’autres formes de rachitisme, et son impact dentaire est sous-estimé.
Le diagnostic repose sur l’association d’une évaluation clinique, biochimique et radiologique :
- Clinique : retard de croissance, douleurs osseuses, déformations des membres, atteintes dentaires.
- Biologie : hypophosphatémie persistante, fuite rénale du phosphate, FGF23 élevé, PTH normale ou légèrement élevée.
- Radiologie : pseudofractures, ostéomalacie, atteinte des métaphyses osseuses.
- Génétique : l’analyse du gène PHEX permet de confirmer l’XLH,
et d’exclure d’autres causes d’hypophosphatémie. En cas d’absence d’analyse moléculaire, l’histoire familiale et des niveaux non supprimés de FGF23 peuvent orienter le diagnostic.
Nouvelles stratégies thérapeutiques : l’impact du burosumab
Le traitement classique repose sur une supplémentation en phosphate oral et vitamine D active, accompagnée d’une surveillance étroite pour prévenir l’hyperparathyroïdie secondaire et la néphrocalcinose.
L’introduction du burosumab, un anticorps monoclonal anti-FGF23, représente une avancée majeure. Il permet de :
- Restaurer la phosphatémie en bloquant l’action excessive de FGF23.
- Améliorer la croissance et la minéralisation osseuse.
- Réduire les douleurs musculosquelettiques et améliorer la mobilité fonctionnelle.
- Diminuer la fréquence des complications dentaires.
Les recommandations actuelles visent à :
- Sélectionner les patients devant débuter ou basculer vers ce traitement.
- Définir la cible optimale des biomarqueurs biologiques (phosphatémie, TmP/GFR).
- Évaluer l’efficacité à long terme sur la croissance, la qualité osseuse, les atteintes neurologiques et orthopédiques.
Quel avenir pour la recherche sur l’XLH ?
Les prochaines étapes incluent :
- Un registre international de patients pour analyser l’histoire naturelle de la maladie et l’efficacité des traitements.
- L’étude des mutations du gène PHEX pour identifier les patients répondant le mieux au burosumab.
- L’amélioration du suivi dentaire et de la prévention des complications bucco-dentaires.
- L’optimisation du dosage et du suivi des patients sous burosumab.
- L’évaluation des traitements en période périnatale et post-chirurgicale.
Améliorer la connaissance de l’XLH est essentielle pour un diagnostic plus précoce et une prise en charge personnalisée. En intégrant ces nouvelles recommandations, en facilitant l’accès au burosumab et en renforçant la prise en charge multidisciplinaire, cela permettra d’améliorer significativement la qualité de vie des patients atteints d’XLH.
Pour en savoir + : Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia.
Haffner D, Emma F, Seefried L, Högler W, Javaid KM, Bockenhauer D, Bacchetta J, Eastwood D, Biosse Duplan M, Schnabel D, Wicart P, Ariceta G, Levtchenko E, Harvengt P, Kirchhoff M, Gardiner O, Di Rocco F, Chaussain C, Brandi ML, Savendahl L, Briot K, Kamenický P, Rejnmark L, Linglart A. Nat Rev Nephrol. 2025 Jan 15.
Hypoparathyroïdie : un pas de plus vers un nouveau traitement
L’essai clinique de phase 3 CALYPSO a franchi une étape clé dans le développement de l’eneboparatide. Cet agoniste du récepteur de la parathormone (PTH) a permis de normaliser les taux de calcium sérique chez des patients atteints d’hypoparathyroïdie chronique tout en réduisant leur dépendance aux suppléments de calcium et de vitamine D actifs.
L’équilibre du métabolisme phosphocalcique en ligne de mire
L’hypoparathyroïdie chronique est une maladie endocrinienne rare qui se traduit par une insuffisance ou une absence de sécrétion de l'hormone parathyroïdienne (PTH) dû à un dysfonctionnement des glandes parathyroïdes. Ce manque de PTH entraîne une hypocalcémie, qui peut être grave et une hyperphosphatémie (excès de phosphate). Jusqu’à présent, les traitements se limitaient à des suppléments en calcium et en vitamine D, sans agir sur la « déficience » hormonale sous-jacente. Récemment, le YORVIPATH (palopegtériparatide) a marqué une avancée significative dans la prise en charge de l’hypoparathyroïdie chronique. Ce traitement est destiné aux adultes dont la maladie reste insuffisamment contrôlée par la supplémentation classique en calcium et en vitamine D, et pour lesquels une substitution hormonale en PTH s’avère nécessaire.
Un essai clinique en deux temps
L’étude CALYPSO repose sur un protocole multicentrique en double aveugle, contrôlé par placebo, impliquant 202 patients répartis en deux groupes. L’objectif : comparer l’efficacité de l’eneboparatide à celle d’un placebo chez des patients sous traitement standard. Le critère principal d’évaluation repose sur la capacité du traitement à restaurer une normocalcémie et à réduire l’usage des suppléments.
Les résultats intermédiaires obtenus après 24 semaines, montrent que l’eneboparatide permet une..
normalisation du calcium sérique, réduisant ainsi la nécessité de suppléments. Le traitement agit directement sur le récepteur PTH1, optimisant l’homéostasie calcique tout en préservant la fonction rénale et osseuse.
Au-delà des marqueurs biologiques, l’essai s’intéresse également à la qualité de vie des patients et aux effets du traitement sur la calciurie, élément clé pour limiter les complications rénales. Tous les participants bénéficieront ensuite d’une phase d’extension à long terme, afin de préciser le profil bénéfice-risque du médicament.
Des perspectives encourageantes
L’eneboparatide a d’ores et déjà obtenu le statut de « fast track » et de médicament orphelin auprès de la Food and Drug Administration (FDA) aux États-Unis et de l’Agence européenne des médicaments (EMA), soulignant l’urgence d’un traitement plus physiologique pour les patients atteints d’hypoparathyroïdie. Les résultats finaux, attendus à 52 semaines, permettront de confirmer son efficacité et sa tolérance.
Si ces données se révèlent concluantes, ce traitement pourrait représenter une avancée majeure, offrant une alternative plus ciblée et mieux adaptée aux besoins des patients, tout en réduisant les risques associés aux thérapies conventionnelles.
APECED : mieux comprendre pour mieux prendre en charge
La polyendocrinopathie auto-immune de type 1, ou syndrome APECED, est une maladie génétique rare due à des mutations bi-alléliques du gène AIRE, associant classiquement une hypoparathyroïdie –une insuffisance surrénalienne – une candidose chronique (infection par champignons).
Une étude nationale menée dans plusieurs centres en France (NCT03751683) vient de livrer ses premiers résultats sur le syndrome APECED. Cette étude a inclus 25 patients issus de 23 familles, permettant d'identifier 11 variants distincts du gène AIRE, responsable de la maladie. Deux variants jamais rapportés auparavant, ont été découverts.
Les patients présentaient en moyenne sept manifestations cliniques distinctes. La triade classique hypoparathyroïdie-insuffisance surrénalienne-candidose chronique était présente chez 19 patients. Parmi les autres symptômes importants : les atteintes pulmonaires, dentaires, digestifs et l’apparition d’autres maladies endocriniennes.
Sur le plan immunologique, l’ensemble des patients possédait des auto-anticorps spécifiques (anti-interféron-α, anti-IL-22 et anti-IL-17F).
Une altération des lymphocytes B, liée à l’âge, a également été observée, soulignant l'importance d'un suivi immunologique régulier.
Ces découvertes confirment que l’APECED est non seulement une maladie rare, mais également sévère, nécessitant un suivi rapproché avec des examens spécifiques systématiques pour permettre un diagnostic précoce. Cela pourrait conduire à une meilleure prise en charge et à une meilleure prévention des complications, notamment par la vaccination pour prévenir les infections et des traitements ciblés.
De nouvelles pistes thérapeutiques, comme l'utilisation récente de médicaments tels que le ruxolitinib, ouvrent également de prometteuses perspectives.
Pour en savoir + : Lessons From Prospective Longitudinal Follow-up of a French APECED Cohort.
Humbert L, Proust-Lemoine E, Dubucquoi S, Kemp EH, Saugier-Veber P, Fabien N, Raymond-Top I, Cardot-Bauters C, Carel JC, Cartigny M, Chabre O, Chanson P, Delemer B, Do Cao C, Guignat L, Kahn JE, Kerlan V, Lefebvre H, Linglart A, Mallone R, Reynaud R, Sendid B, Souchon PF, Touraine P, Wémeau JL, Vantyghem MC. J Clin Endocrinol Metab. 2025 Feb 18;110(3):e757-e773.
L’hypophosphatasie : une charge de morbidité sous-estimée
L’hypophosphatasie (HPP) est une maladie génétique rare caractérisée par une déficience en phosphatase alcaline non spécifique des tissus (TNAP), entraînant des atteintes osseuses, dentaires, musculaires et neurologiques. Si les formes les plus sévères apparaissent dès la petite enfance, les manifestations peuvent varier considérablement selon les individus. Une question reste en suspens : le nombre de mutations du gène ALPL influence-t-il la gravité de la maladie ?
Première analyse comparative entre patients avec une ou plusieurs mutations
Pour mieux comprendre cette corrélation, une étude menée à partir du Global HPP Registry a analysé 685 patients atteints d’HPP. L’objectif : comparer les caractéristiques cliniques et fonctionnelles des patients ayant une mutation (hétérozygotes) ou deux mutations (homozygotes ou hétérozygotes composites) du gène ALPL.
Sur les 685 patients inclus, 82,9 % présentaient une seule mutation ; 16,9 % avaient deux mutations ; un patient possédait trois mutations, un cas rarissime.
Une atteinte plus sévère avec plusieurs mutations
Les patients ayant deux ou plusieurs mutations présentaient un taux plus élevé de manifestations osseuses (52,1 % vs 32,6 %), dentaires (73,5 % vs 56 %), musculaires (36,8 % vs 23,6 %) et neurologiques (22,2 % vs 8,8 %).
L’analyse du test de marche de 6 minutes a révélé des résultats similaires entre les enfants des deux groupes. En revanche, chez les adultes, ceux ayant deux mutations ou plus parcouraient en moyenne 293 mètres contre 466 mètres pour ceux avec une seule mutation.
Fait surprenant : les scores des échelles de qualité de vie ne montraient pas de différence majeure entre les groupes. Cela suggère que d’autres facteurs, comme les stratégies d’adaptation, le suivi médical et le soutien familial, influencent fortement la perception de la maladie.
Vers une prise en charge plus adaptée
Cette étude met en lumière une hétérogénéité importante des manifestations de l’hypophosphatasie, qui ne peut être expliquée uniquement par le nombre de mutations du gène ALPL. Elle souligne également l’importance d’une approche individualisée pour améliorer le diagnostic et la prise en charge des patients.
Il s’agit de la première étude évaluant la différence de charge de morbidité entre les patients ayant un seul variant ALPL et ceux présentant deux variants ou plus. Elle suggère l’importance d’approfondir les interactions génétiques et biologiques qui influencent l’évolution de la maladie. Des pistes comme les biomarqueurs de sévérité ou de nouvelles cibles thérapeutiques sont à l’étude pour optimiser la prise en charge. L’hypophosphatasie est une maladie aux multiples visages… et le séquençage génétique n’a pas encore livré tous ses secrets !
Pour en savoir + : Disease burden by ALPL variant number in patients with non-life-threatening hypophosphatasia in the Global HPP Registry.
Kishnani PS, Seefried L, Dahir KM, Martos-Moreno GÁ, Högler W, Greenberg CR, Fang S, Petryk A, Mowrey WR, Linglart A, Ozono K.J Med Genet. 2025 Feb 18:jmg-2024-110383.
Vers une thérapie génique pour l’hypophosphatasie ?
L’hypophosphatasie (HPP) est une maladie génétique rare causée par un déficit en phosphatase alcaline non spécifique des tissus (TNAP). Cette enzyme joue un rôle essentiel dans la minéralisation des os et des dents. Jusqu’ici, les traitements disponibles se limitaient essentiellement à l’enzymothérapie substitutive. Une nouvelle étude explore une approche innovante : la thérapie génique par vecteur viral, visant à restaurer durablement l’expression de TNAP.
Une injection unique pour restaurer l’enzyme manquante
L’équipe de recherche a développé un vecteur viral (de type AAV8) capable d’amener le gène de la TNAP directement dans l’organisme. Une seule injection de ce vecteur – appelé AAV8-TNAP-D10 – a déjà permis, dans de précédents travaux, de corriger les anomalies osseuses et dentaires chez des souris atteintes d’HPP.
Dans cette nouvelle étude, les chercheurs ont testé plusieurs doses de ce traitement sur deux modèles de la maladie :
- Modèle d’HPP infantile (Alpl-/-) : injection intramusculaire à 4 jours de vie.
- Modèle d’HPP d’apparition tardive (AlplPrx1/Prx1) : injection unique à 8 semaines d’âge.
Des résultats encourageants… mais à moduler
Les résultats ont montré une amélioration progressive selon la dose administrée :
- Une augmentation de l’activité TNAP dans le sang.
- Une réduction des taux de pyrophosphate, une substance qui freine la minéralisation osseuse.
- Une correction complète du squelette chez les jeunes souris traitées à la dose la plus élevée.
Cependant, les effets varient selon l’âge et le sexe des souris :
- Chez les femelles, la réponse au traitement est meilleure, avec une correction quasi complète de l’atteinte osseuse.
- Chez les mâles, l’efficacité est plus faible :
la protéine réparatrice se retrouve en quantité plus importante dans le foie, et non dans les os, ce qui limite son effet. De plus, une augmentation des marqueurs inflammatoires a été observée chez les mâles traités, ce qui pourrait être un frein à l’efficacité du traitement.
Des effets secondaires à surveiller ?
Bien que la thérapie génique soit globalement bien tolérée, des calcifications ectopiques ont été détectées dans les tissus mous à des doses élevées. Ces effets secondaires, bien que rares, rappellent l’importance d’un suivi médical attentif si ce traitement venait à être proposé à l’homme.
Les prochaines étapes
Ces résultats valident l’efficacité de la thérapie génique dans l’HPP, mais soulignent également la nécessité d’adapter les doses et de mieux comprendre les différences biologiques entre les sexes et les âges.
Les chercheurs envisagent maintenant :
- D’optimiser le ciblage du traitement pour éviter l’accumulation dans le foie.
- De mener des études à plus long terme pour évaluer la sécurité et la durabilité du traitement.
- Et de préparer des essais cliniques chez les patients atteints d’HPP.
En conclusion, la thérapie génique pour l’HPP progresse vers la réalité clinique, mais plusieurs ajustements sont nécessaires avant de garantir une efficacité optimale pour tous les patients.
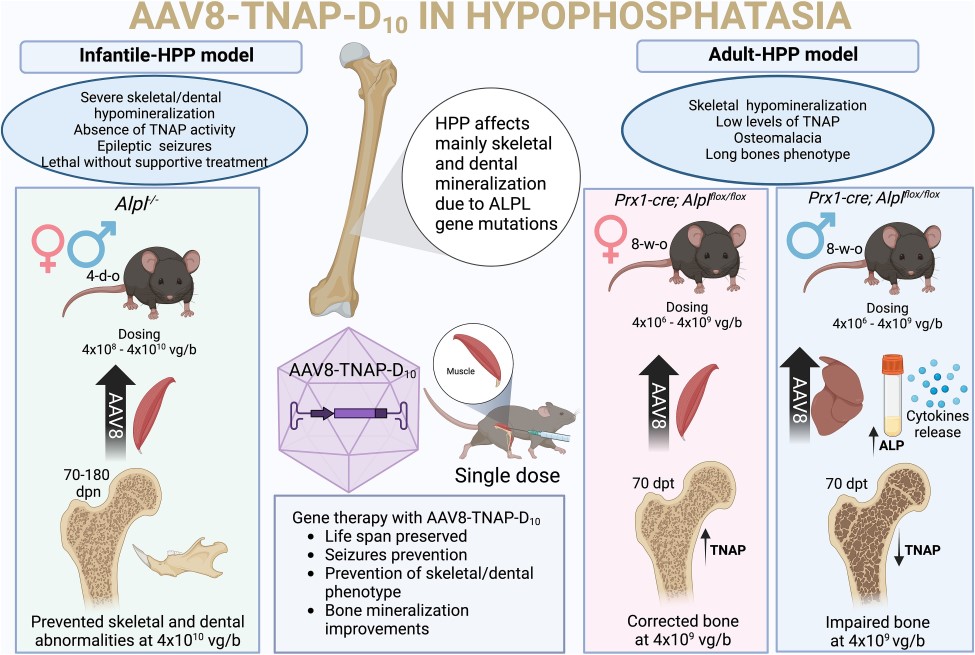
Figure tirée ©de Oliveira FA et al. 2025. Published by Oxford University Press on behalf of the American Society for Bone and Mineral Research, https://doi.org/10.1093/jbmr/zjaf005
Pour en savoir + : Preclinical evaluation of the efficacy and safety of AAV8-TNAP-D10 in Alpl-/- and AlplPrx1/Prx1 mouse models for the treatment of early and late-onset hypophosphatasia.
de Oliveira FA, Tokuhara CK, Mohamed FF, Narisawa S, Lira Dos Santos EJ, Andras NL, Shadid M, Miyake K, Foster BL, Millán JL.J Bone Miner Res. 2025 Jan 12:zjaf005.


