Actu - septembre 2024

Sommaire
🔎 Qu’en est-il du burosumab chez les nourrissons de moins d’un an atteints d’XLH ?
🔎 Lien entre l’XLH et l'obésité : ce qu'il faut savoir
🔎 Pas de risque cardiovasculaire accru chez les adultes XLH sous traitement standard !
🔎 L’énéboparatide, un agoniste du PTHR1, prometteur dans le traitement de l'hypoparathyroïdie
🔎 Hypophosphatasie : déchiffrer les variants du gène ALPL
🔎 De nouvelles révélations du registre mondial de l’HPP sur son paysage génétique !
🔎 L’efficacité de l'asfotase alfa dans le traitement de l’hypophosphatasie chez l’adulte
Qu’en est-il du burosumab chez les nourrissons
de moins d’un an atteints d’XLH ?
L'hypophosphatémie liée à l'X (XLH) est une maladie génétique rare caractérisée par des niveaux élevés de FGF23 (facteur de croissance des fibroblastes 23) dans le sang, ce qui entraîne une diminution des taux de phosphate dans le sang et une réduction des niveaux sériques de vitamine D. Il s’agit d’une maladie génétique, dominante liée au chromosome X, qui est la cause la plus fréquente des anomalies génétiques du rachitisme.
Le burosumab, un anticorps monoclonal entièrement humain, cible et neutralise le FGF23, permettant ainsi de rétablir des niveaux normaux de phosphate et d'améliorer les anomalies osseuses associées à la maladie. Depuis 2018, le burosumab est autorisé pour le traitement de l’XLH avec signes radiographiques d’atteinte osseuse chez les enfants âgés d’un an et plus et chez les adolescents en phase de croissance osseuse.
Mais qu'en est-il des nourrissons de moins d'un an ?
L'étude ouverte de phase 1/2 BUR-CL207 a récemment examiné la sécurité, la tolérance et l'efficacité du burosumab dans cette
tranche d'âge. Les résultats, présentés lors de l'International Conference on Children’s Bone Health en juin dernier, ont inclus 16 nourrissons répartis en trois groupes : ceux de moins de 6 mois recevant une dose initiale de 0,4 mg/kg, et ceux de 6 à moins de 12 mois recevant des doses de 0,4 mg/kg ou 0,8 mg/kg.
Les résultats montrent que le burosumab est généralement bien toléré. Les effets indésirables liés au traitement incluent une augmentation de l'hormone parathyroïdienne et, dans un cas rare, une crâniosynostose (une anomalie du crâne). Ces effets indésirables n'ont pas entraîné d'arrêt du traitement. Le burosumab a également été efficace pour améliorer les niveaux de phosphate sérique, de 1,25-dihydroxyvitamine D et de phosphatase alcaline. De surcroît, les résultats radiographiques ont démontré une amélioration des signes cliniques de rachitisme. Ces résultats sont prometteurs pour le développement osseux et la croissance des nourrissons atteints de XLH.
Pour en savoir + : An open-label, multicentre, non-randomised study investigating the safety, tolerability and efficacy of burosumab in infants with X-linked hypophosphataemia: Results from the BUR-CL207 study.
Linglart A, Emma F, Bacchetta J, and al. ICCBH, OC 6.2, juin 2024.
Lien entre l’XLH et l'obésité : ce qu'il faut savoir
L'hypophosphatémie liée à l’X (XLH) est-elle réellement associée à un risque accru d'obésité et de complications métaboliques ?
C'est la question que soulève une nouvelle étude menée par le Dr Anne Lise LECOQ, le Pr Peter KAMENICKÝ et leurs collègues, du centre de référence coordonnateur des maladies rares du métabolisme du calcium et du phosphate (CaP), à l’hôpital Bicêtre (AP-HP), laboratoire U1185 INSERM/Université Paris-Saclay.
Dans cette étude publiée récemment dans l’European Journal of Endocrinology, les chercheurs ont évalué la prévalence du surpoids et de l’obésité chez les adultes atteints de XLH. Ils ont évalué la proportion de patients ayant un indice de masse corporelle (IMC) supérieur à 25 kg/m², ainsi que des critères secondaires comme le taux de graisse corporelle et les surfaces des tissus adipeux.
Les résultats ont révélé que 56 % des participants étaient en surpoids ou obèses, avec un IMC médian de 25,3 kg/m². L’IMC était lié au taux de masse grasse et aux surfaces des tissus adipeux sous-cutanés et intra-abdominaux. Cependant, les chercheurs ont noté que les troubles du métabolisme du glucose (telles que l’altération de la glycémie à jeun, l’intolérance au glucose, et le diabète) ne sont pas plus fréquents chez les patients XLH que chez les témoins.
Ces résultats suggèrent que, bien que les patients XLH présentent une quantité importante de masse grasse, ils ne montrent pas nécessairement de perturbations métaboliques graves. Toutefois, il est particulièrement important de surveiller les patients avec une obésité sévère (classe III), car un excès de poids favorise les complications ostéoarticulaires et aggrave la maladie. Les chercheurs expriment leur intérêt pour de futures études visant à explorer davantage ces données et à déterminer si elles sont liées à des mécanismes particuliers de l’XLH.
Pour en savoir + : Metabolically healthy obesity in adults with X-linked hypophosphatemia
Lecoq AL, Schilbach K, Rocher L, Trabado S, Briot K, Herrou J, Forbes A, Garnier A, Piketty M, Bidlingmaier M, Rothenbuhler A, Linglart A, Carette C, Chaumet-Riffaud P, Kamenický P.Eur J Endocrinol. 2024 Aug 5;191(2):156-165. doi: 10.1093/ejendo/lvae089.
Pas de risque cardiovasculaire accru chez les adultes XLH sous traitement standard !
Bien que des études aient établi un lien direct entre le FGF23 et la mortalité cardiovasculaire chez les patients atteints d'insuffisance rénale chronique, les données sur l'impact cardiovasculaire chez les patients atteints de XLH sont limitées et parfois contradictoires.
Pour mieux comprendre ces effets, les équipes de néphrologie et de rhumatologie de l'Hôpital Edouard Herriot à Lyon au sein du centre de référence constitutif des maladies rares du métabolisme du calcium et du phosphate (CRMR CaP) aux hospices civils de Lyon (HCL) ont mené une étude observationnelle auprès de vingt-deux patients adultes atteints de XLH. Les cliniciens ont principalement évalué la fréquence de l'hypertrophie ventriculaire gauche (HVG) et la présence de l’hypertension artérielle (HTA). En parallèle, deux autres patients du London Health Sciences Centre Renal Program, Ontario, Canada, ont participé à l'étude et ont subi une imagerie par résonance magnétique (IRM), afin de mesurer la concentration de sodium dans les tissus avant et après l'injection du burosumab, un médicament utilisé dans le traitement de l’XLH (NCT03004547).
Les résultats montrent que la majorité des patients avaient des niveaux normaux de phosphate et de FGF23 dans le sang. La pression artérielle moyenne était de 124/68 mm Hg, avec seulement 9 % des patients ayant une hypertension artérielle. En outre, 69 % des patients n'ont pas développé de complications cardiaques. Les examens par IRM n'ont révélé aucun changement significatif dans le contenu en sodium des tissus avant et après le traitement par burosumab chez les deux patients évalués.
Les auteurs rapportent que les patients atteints de XLH, suivis de manière précoce et rigoureuse, ne semblent pas présenter un risque accru de développer de l'hypertension artérielle ou une hypertrophie ventriculaire gauche, malgré les anomalies associées au FGF23.
Ces résultats encouragent la poursuite des études pour confirmer ces observations et affiner les stratégies thérapeutiques pour le XLH.
Pour en savoir + : Reassuring Data on the Cardiovascular Risk in Adults With X-linked Hypophosphatemia Receiving Conventional Therapy.
Bouzemane A, Vignot E, Derain Dubourg L, De Mul A, Molin A, Chapurlat R, Fontanges E, Delsart D, Akbari A, Huang SHS, McIntyre CW, Bacchetta J, Lemoine S. J Clin Endocrinol Metab. 2024 Jan 18;109(2):e488-e494.
L’énéboparatide, un agoniste du PTHR1, prometteur dans le traitement de l'hypoparathyroïdie
L'hypoparathyroïdie (HP) est une maladie rare, caractérisée par un déficit de sécrétion de l'hormone parathyroïdienne (PTH), entraînant des niveaux anormalement bas de calcium dans le sang (hypocalcémie), des niveaux anormalement élevés de phosphate dans le sang (hyperphosphatémie) et une augmentation de l'excrétion de calcium dans l'urine (hypercalciurie). Cette maladie touche principalement les femmes âgées de 50 ans et plus.
Le traitement standard actuel, basé sur des doses élevées de suppléments en calcium et en vitamine D, ne parvient souvent pas à normaliser les niveaux de calcium sanguin. De plus, il peut aggraver l'hypercalciurie, entraînant un dysfonctionnement des reins et une insuffisance rénale progressive due à des dépôts de calcium et des calculs rénaux. Face à ces limites, l'idée de
remplacer directement l'hormone manquante, la PTH, a émergé.
Une récente étude internationale de phase II, dont les résultats ont été publiés dans The Journal of Clinical Endocrinology & Metabolism, a examiné les effets de l'énéboparatide (AZP-3601) dans le traitement de l'hypoparathyroïdie. Ce peptide thérapeutique expérimental se lie avec une haute affinité à une conformation spécifique du récepteur de la PTH pour induire un effet prolongé sur le métabolisme du calcium et contrôler ainsi les symptômes de l’hypoparathyroïdie. De plus, son action peut limiter l’excrétion urinaire du calcium en restaurant la réabsorption de calcium par le rein, dans le but de prévenir le déclin progressif des fonctions rénales et le développement de maladies rénales chroniques. Le traitement est administré en injections sous-cutanées quotidiennes.
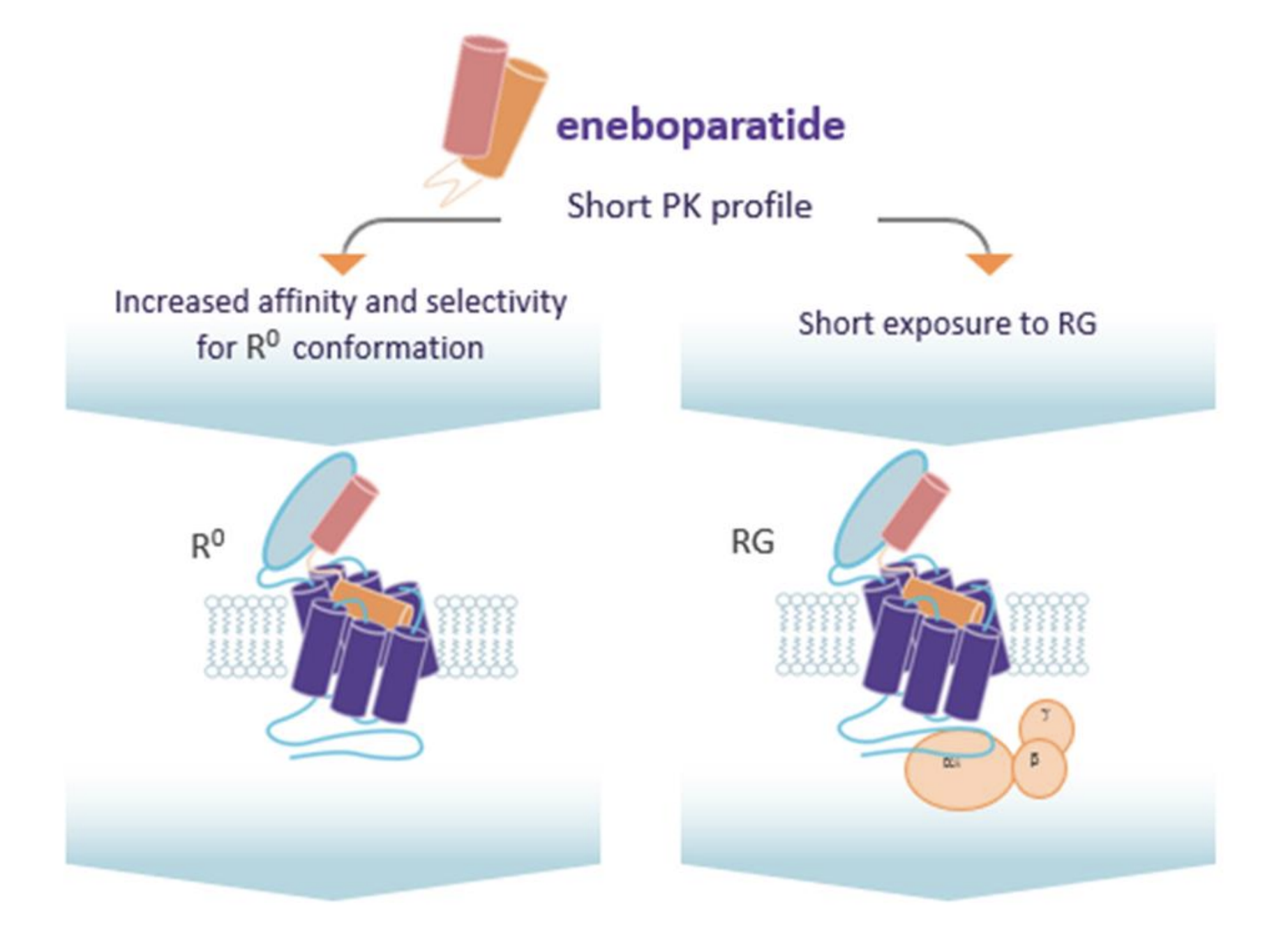
Mécanisme d'action de l'énéboparatide
(Figure tirée de la communication de Lucile Figueres et al. ENDO 2023)
Le récepteur de la PTH existe sous 2 conformations allostériques principales : R0 et RG. Le récepteur est sous la conformation R0 lorsqu’il est lié à la molécule au niveau du tubule rénal. Ceci permet une endocytose de l’AZP-3601 et une activation prolongée de la voie de signalisation AMPc/PKA par cycles multiples et donc une réabsorption tubulaire durable de calcium. Au niveau de l’os, le récepteur est sous sa conformation RG en présence de la molécule. L’effet est donc moins prolongé.
Les résultats de l'étude ont démontré qu'après trois mois de traitement, l'énéboparatide a réussi à maintenir les niveaux de calcium sanguin dans la fourchette cible pour la plupart des patients. En outre, 92 % des patients ont pu arrêter leur
traitement conventionnel.
Le médicament a également entraîné une augmentation équilibrée des biomarqueurs osseux, reflétant un renouvellement osseux plus physiologique, et a rapidement normalisé l'excrétion urinaire de calcium, même chez les patients présentant une hypercalciurie. Le traitement a été bien tolérée, sans effets indésirables graves signalés.
L’étude CALYPSO, de phase III multicentrique, randomisée, contrôlée par placebo, en double aveugle est actuellement en cours pour évaluer davantage ce traitement, avec des résultats attendus en 2025.
Pour en savoir + : An Open-label Phase 2 Study of Eneboparatide, a Novel PTH Receptor 1 Agonist, in Hypoparathyroidism
Takacs I, Mezosi E, Soto A, Kamenický P, Figueres L, Galvez Moreno MA, Lemoine S, Borson-Chazot F, Capel I, Ouldrouis T, Lucas N, Allas S, Sumeray M, Ovize M, Mannstadt M. J Clin Endocrinol Metab. 2024 Mar 6:dgae121. doi: 10.1210/clinem/dgae121.
HPP : déchiffrer les variants du gène ALPL
Pour mieux comprendre l’influence de la génétique sur le risque de développer ou non une maladie, les généticiens du monde entier cherchent à identifier les variants génétiques associés aux maladies humaines et aux phénotypes résultant de la combinaison de plusieurs gènes. Un variant génétique peut être décrit comme toute différence par rapport à ce qui est considéré comme une séquence normale d’ADN chez l’humain. Un variant peut être interprété comme étant bénin, potentiellement bénin, probablement pathogène, pathogène, ou encore de signification inconnue (VSI) en raison de données insuffisantes.
L'hypophosphatasie (HPP) est une maladie génétique rare causée par des variants pathogènes dans le gène ALPL, qui code pour l’enzyme phosphatase alcaline non spécifique des tissus (ALP). Le diagnostic de l'HPP est souvent retardé, en partie en raison de la grande variabilité des symptômes cliniques, qui peuvent apparaître à tout âge et varier considérablement d'une personne à l'autre, même au sein d'une même famille.
Il est donc important de mieux comprendre le spectre génétique et phénotypique de cette maladie. L'évaluation et la reclassification approfondies des VSI constituent une étape essentielle à cet égard, non seulement pour mieux identifier les formes les plus bénignes de l'HPP, mais aussi pour caractériser les phénotypes nouveaux. Cela permet d'apporter plus de certitude dans le diagnostic et d'améliorer la prise en charge clinique des patients. C'est dans ce but que la base de données
de variants génétiques ALPL (https://alplmutationdatabase.jku.at/) a été créée.
Le projet de classification des variants du gène ALPL a été établi pour reclassifier les VSI et mettre à jour en continu les informations génétiques, phénotypiques et fonctionnelles des variants dans la base de données. Ce projet propose un système unique pour les cliniciens, généticiens, conseillers en génétique et chercheurs, leur permettant de soumettre des VSI pour classification. Un consortium international et multidisciplinaire d'experts en HPP a été formé pour reclassifier les VSI, en suivant un processus rigoureux conforme aux directives de classification des variants de l'ACMG/AMP*. Celui-ci inclut une évaluation du phénotype clinique, une recherche approfondie de la littérature utilisant des technologies d'intelligence artificielle, une évaluation génétique moléculaire et des tests fonctionnels in vitro des variants dans un modèle de co-transfection pour mesurer l'activité résiduelle de l'ALP.
La mise en place d’une méthode efficace pour classer ces VSI constitue une avancée majeure pour le conseil génétique et la prise de décision médicale pour les patients et les familles. Le projet pourrait également servir de référence pour la collaboration multidisciplinaire dans l'interprétation des VSI d'autres maladies rares.
*ACMG (American College of Medical Genetics and Genomics)/ AMP (Association for Molecular Pathology)
Pour en savoir + : The Global ALPL gene variant classification project: Dedicated to deciphering variants
Farman MR, Rehder C, Malli T, Rockman-Greenberg C, Dahir K, Martos-Moreno GÁ, Linglart A, Ozono K, Seefried L, Del Angel G, Webersinke G, Barbazza F, John LK, Delana Mudiyanselage SMA, Högler F, Nading EB, Huggins E, Rush ET, El-Gazzar A, Kishnani PS, Högler W.Bone. 2024 Jan;178:116947. doi: 10.1016/j.bone.2023.116947.
De nouvelles révélations du registre mondial de l’HPP sur son paysage génétique !
L’HPP est caractérisée par une grande variabilité clinique, influencée par des facteurs génétiques et environnementaux. Comprendre comment les variants génétiques de l'HPP sont répartis selon les régions géographiques, les groupes ethniques et les âges des patients pourrait fournir des informations essentielles pour mieux diagnostiquer et comprendre cette maladie rare.
À cette fin, des chercheurs ont analysé les différents variants génétiques associés à l'HPP à partir des données du registre mondial de l’HPP.
Au total, 814 patients ont été inclus dans l'analyse. Ils avaient un diagnostic confirmé d'HPP, une faible activité de l'enzyme phosphatase alcaline dans le sang, et au moins un variant génétique. Les patients étaient originaires d'Europe (48,9 %), d'Amérique du Nord (36,7 %), du Japon (10,2 %), d'Australie (2,6 %) et d'autres régions (1,6 %). La majorité (74,7 %) avait un seul variant génétique dans le gène ALPL, tandis que 25,3 % en
avaient deux ou plus. La grande majorité des variants génétiques observés étaient connus pour provoquer la maladie (95,6 %), tandis que 4,4 % étaient encore incertains.
Les résultats étaient généralement similaires d'une région à l'autre, sauf au Japon où une proportion plus élevée de patients (68,7 %) avait deux variants génétiques ou plus. Cela pourrait être dû à une apparition plus précoce de la maladie avant l'âge de 6 mois au Japon (53 % contre 10 à 23 % ailleurs). Les mutations par décalage du cadre de lecture et les délétions étaient aussi plus fréquentes au Japon. De plus, 23 nouveaux variants génétiques ont été découverts, principalement en Europe.
Cette étude a permis de confirmer les variants génétiques déjà connus et contribue à mieux comprendre la diversité génétique de l'HPP selon les régions géographiques, facilitant ainsi le développement de stratégies de diagnostic et de traitement plus ciblées pour les patients.
Pour en savoir + : New insights into the landscape of ALPL gene variants in patients with hypophosphatasia from the Global HPP Registry
Kishnani PS, Seefried L, Dahir KM, Martos-Moreno GÁ, Linglart A, Petryk A, Mowrey WR, Fang S, Ozono K, Högler W, Rockman-Greenberg C.Am J Med Genet A. 2024 Jun 17:e63781. doi: 10.1002/ajmg.a.63781.
L’efficacité de l'asfotase alfa dans le traitement de l’HPP chez l’adulte
L'hypophosphatasie (HPP) est une maladie génétique rare du métabolisme osseux caractérisée par un défaut d’activité enzymatique de la phosphatase alcaline (ALP), qui affecte la minéralisation des os et des dents. Chez l’adulte, les symptômes de cette maladie peuvent être très divers et surtout invalidants : douleurs osseuses, retard moteur avec des difficultés physiques, fatigue, insuffisance musculaire, pertes de dents prématurées, etc., ce qui contribue à une diminution de la qualité de vie des patients. L’HPP peut être traitée par un substitut enzymatique, l’asfotase alfa, bien que les données en vie réelle chez l’adulte soient limitées.
Dans cette étude publiée récemment dans l’Orphanet Journal of Rare Diseases, les auteurs rapportent les résultats de l'efficacité de l'asfotase alfa à travers des mesures fonctionnelles et des résultats rapportés par les patients (PRO) dans une cohorte
de 190 patients adultes inscrits dans le registre mondial de l’HPP (le plus grand étudié à ce jour) répondant aux critères d'inclusion.
Les résultats montrent que l'asfotase alfa est associée à des améliorations de la mobilité, des mesures de la qualité de vie et de la douleur dès 6 mois de traitement, des bénéfices qui se maintiennent sur trois ans de suivi. Ces résultats sont particulièrement importants pour les patients souffrant de douleurs chroniques et de limitations fonctionnelles en raison de cette maladie rare. Bien que des réactions au site d'injection aient été signalées, le traitement a été généralement bien toléré.
Ces résultats positifs renforcent l'importance du traitement par asfotase alfa pour améliorer la vie quotidienne des personnes atteintes d'HPP.
En savoir + : Effectiveness of asfotase alfa for treatment of adults with hypophosphatasia: results from a global registry
Kishnani PS, Martos-Moreno GÁ, Linglart A, Petryk A, Messali A, Fang S, Rockman-Greenberg C, Ozono K, Högler W, Seefried L, Dahir KM. Orphanet J Rare Dis. 2024 Mar 8;19(1):109. doi: 10.1186/s13023-024-03048-6.


