Actus - septembre 2024

Sommaire
🔎 L’ostéopétrose et maladies associées chez l’adulte : ce que l’on sait et ce qui reste à faire !
🔎 L’ostéogenèse imparfaite : que révèlent les premiers résultats de l’enquête IMPACT ?
🔎 L'achondroplasie : de nouvelles perspectives thérapeutiques prometteuses
🔎 Optimiser les RDV médicaux avec le nouveau guide pratique pour les adultes atteints d'achondroplasie
🔎 La greffe de moelle osseuse allogénique : le coup de boost dans la dysplasie cranio-métaphysaire
L’ostéopétrose et maladies associées chez l’adulte : ce que l’on sait et ce qui reste à faire !
À la suite d'un travail collaboratif réalisé par les experts de l’European Calcified Tissue Society (ECTS) et de l’ERN BOND, sous la direction du Pr Martine COHEN-SOLAL, responsable du centre de référence constitutif des maladies osseuses constitutionnelles (MOC) à l’hôpital Lariboisière (AP-HP), une récente revue de littérature internationale publiée dans l’European Journal of Medical Genetics présente en détail les caractéristiques cliniques principales de l'ostéopétrose chez l’adulte, sa prise en charge, et offre des perspectives sur les axes de recherche.
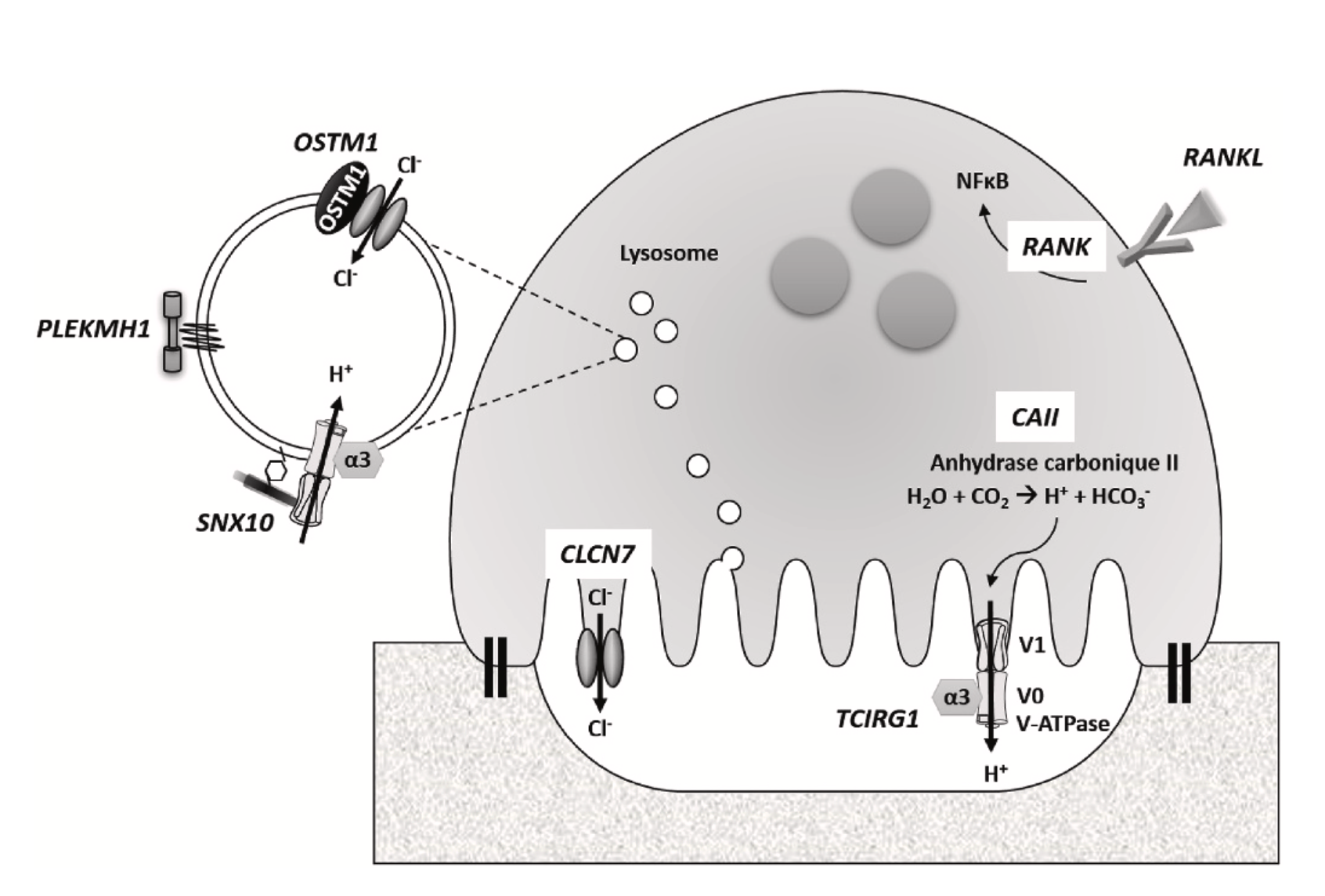
L’os est un tissu en perpétuel renouvellement, et l’homéostasie osseuse dépend de l’équilibre d’action de deux types cellulaires : les ostéoblastes, responsables de la formation osseuse, et les ostéoclastes, chargés de la résorption. L’ostéopétrose se réfère à un groupe d'anomalies osseuses rares et héréditaires qui résulte d’un défaut de la résorption par les ostéoclastes et qui aboutit à une hyperminéralisation du squelette entraînant un risque accru de fractures, notamment des os longs.
Une nouvelle classification
L'ostéopétrose a été décrite pour la première fois par ALBERS-SCHÖNBERG en 1904. Au fil du temps, plusieurs formes d'ostéopétrose ont été distinguées : les formes autosomiques récessives (ARO), qui sont sévères et diagnostiquées dans les premières années de vie, la forme intermédiaire, la forme liée à l'X et la forme autosomique dominante (ADO), de gravité variable, diagnostiquée pendant l'adolescence ou au début de l'âge adulte.
Les récentes avancées génétiques ont permis une nouvelle classification des sous-types d'ostéopétrose, selon le
génotype et les autres maladies à densité osseuse élevée (les maladies osseuses sclérosantes héréditaires rares). Dans les formes ARO, 13 entités distinctes sont répertoriées, résultant de variations pathogènes dans 11 gènes différents. La forme la plus sévère d'ARO (appelée ostéopétrose infantile maligne (MIOP)), diagnostiquée peu après la naissance est fatale si elle n'est pas prise en charge.
L'ostéopétrose autosomique dominante (anciennement ADO type 2), également connue sous le nom de maladie d'Albers-Schönberg, est la forme la plus fréquente observée principalement chez les adultes, avec une incidence estimée à 1 naissance sur 20 000. La majorité des cas d’ADO sont associés à des mutations dans le gène CLCN7, codant pour le canal chlorure 7, qui joue un rôle important dans la régulation de l'équilibre du chlore dans les cellules osseuses. À ce jour, plus de 34 mutations de CLCN7 ont été répertoriées. La pénétrance de la maladie d'Albers-Schönberg est incomplète, puisqu'environ 66 % des patients porteurs de la mutation présentent des manifestations cliniques, avec une variabilité d’expression considérable, même parmi les membres d'une même famille.
Les principales mutations génétiques impliquées dans les ostéopétroses
(Figure tirée de l’article Funck-Brentano T, et al. Eur J Med Genet. 2024)
L’ADO présente un large éventail de symptômes, allant des simples anomalies radiologiques à un handicap sévère dû à des complications qui peuvent inclure des fractures fréquentes, souvent atypiques (situées au niveau de la métaphyse ou de la diaphyse), une infection de l'os (ostéomyélite) avec des abcès dentaux ou caries, ainsi que des complications hématologiques. Une complication rare mais grave de la maladie est la compression des nerfs crâniens qui peut occasionner une perte auditive et visuelle.
Une prise en charge multidisciplinaire
En raison de la diversité des complications affectant plusieurs organes, la prise en charge des patients adultes atteints d'ostéopétrose nécessite une approche multidisciplinaire, idéalement dans des centres de référence ou de compétence des maladies rares osseuses. En dehors de la transplantation de cellules souches hématopoïétiques pour certaines formes infantiles, le traitement des patients atteints d'ostéopétrose n'a pas été standardisé et vise principalement à prévenir ou à traiter les complications de la maladie.
Un traitement innovant devrait induire la capacité des ostéoclastes à résorber l'os. Des approches originales utilisant la thérapie génique ou à base d'ARN sont en cours d’étude et devraient être transférées à la clinique dans un proche avenir.
Les orientations futures pour la recherche
Cependant, il persiste un important besoin de recherche pour combler les lacunes de connaissances sur l'histoire naturelle de la maladie, les résultats rapportés par les patients (PRO) et les
options thérapeutiques actuellement limitées. Dans cette perspective, les experts ont identifié plusieurs domaines clés nécessitant des recherches supplémentaires pour mieux appréhender et traiter l'ostéopétrose chez les adultes:
- La compréhension de l'histoire naturelle et épidémiologie : il est crucial de mieux appréhender la prévalence, les caractéristiques et l'évolution des fractures, etc. En effet, des études explorant les changements de densité osseuse et de biomarqueurs osseux au fil du temps en relation avec le génotype sont nécessaires. Cela pourrait aider à expliquer la variabilité du phénotype chez les patients.
- L’amélioration de la collecte de données et de la collaboration internationale : étant donné le caractère rare de l'ostéopétrose, une collaboration internationale et le partage des données au sein d'un consortium permettraient d'obtenir une vision plus complète de la maladie.
- L’élaboration de recommandations pour la prise en charge orthopédique : le développement de directives et le partage d'expériences entre chirurgiens orthopédiques spécialisés sont essentiels pour optimiser la prise en charge des fractures et des remplacements articulaires chez les patients atteints d'ostéopétrose.
- L’évaluation neurologique (anomalies d'imagerie, fonctions cognitives) : elle est nécessaire pour le suivi des patients et permettra d'identifier les critères pour repérer les patients à risque de complications et de les sélectionner pour de futurs traitements.
En outre, plusieurs questions nécessitent des approches de recherche fondamentale ou translationnelle, notamment le mécanisme de la douleur dans les os ou les articulations, la vascularisation de l'os, la variabilité du phénotype en relation avec la défaillance des ostéoclastes, les effets de la transplantation de moelle osseuse, etc.
Pour en savoir + : Osteopetrosis and related osteoclast disorders in adults: A review and knowledge gaps On behalf of the European calcified tissue society and ERN BOND
Funck-Brentano T, Zillikens MC, Clunie G, Siggelkow H, Appelman-Dijkstra NM, Cohen-Solal M. Eur J Med Genet. 2024 Apr 7;69:104936.
L’ostéogenèse imparfaite : que révèlent les premiers résultats de l’enquête IMPACT ?
L’ostéogenèse imparfaite (OI), ou maladie des os de verre, est une maladie génétique rare du tissu conjonctif caractérisée par une fragilité osseuse et une faible masse osseuse. Elle concerne entre 1 et 5 personnes sur 10 000. Actuellement, aucun traitement curatif n'est disponible pour cette maladie rare. Les interventions médicales visent principalement à gérer et à réduire les fractures, les douleurs et les déformations osseuses, tout en favorisant la mobilité des patients.
Alors que certains aspects de l'OI et leur impact sur la qualité de vie liée à la santé sont bien étudiés, d'autres restent mal compris. C’est dans cette optique que l’enquête IMPACT (Living with osteogenesis imperfecta: understanding experiences based on community insight & evidence) a été lancée pour mieux comprendre l'impact humain, clinique et économique de l'OI sur les individus malades, leurs familles, leurs aidants et la société, en général.
Cette enquête internationale en ligne a été élaborée en
collaboration avec des chercheurs et des membres des associations de patients (OIFE - Osteogenesis Imperfecta Federation Europe, OI Foundation, etc.) afin d'assurer une conception pertinente et inclusive. Traduite en huit langues (anglais, allemand, italien, néerlandais, français, russe, espagnol et portugais), elle a été menée de juillet à septembre 2021, et a ciblé les adultes (âgés de ≥ 18 ans) ou les adolescents (âgés de ≥ 12 à 17 ans) atteints d'OI, ainsi que les aidants et les autres proches.
L'enquête IMPACT a recueilli 2208 questionnaires, portant sur 2988 individus, dont 2312 étaient atteints d’OI. Elle révèle que, quel que soit l’âge, les personnes atteintes d'OI rencontrent de nombreux symptômes évolutifs qui affectent leur qualité de vie, avec une présence constante de la douleur et de la fatigue. Les analyses à venir permettront d'approfondir les connaissances sur l'impact économique, le parcours de soins et le bien-être des aidants, dans le but d'améliorer les traitements et les soins pour la communauté des personnes atteintes de cette maladie rare.
Pour en savoir + : The IMPACT survey: a mixed methods study to understand the experience of children, adolescents and adults with osteogenesis imperfecta and their caregivers.
Westerheim I, Hart T, van Welzenis T, Wekre LL, Semler O, Raggio C, Bober MB, Rapoport M, Prince S, Rauch F. Orphanet J Rare Dis. 2024 Mar 21;19(1):128. doi: 10.1186/s13023-024-03126-9.
L'achondroplasie : de nouvelles perspectives thérapeutiques prometteuses
L’achondroplasie (ACH) désigne la forme de nanisme la plus courante. Elle concerne environ une naissance sur 25 000. Le trait principal de cette maladie est une taille plus petite que la moyenne, associée à un raccourcissement des membres.
C’est en 1994 que le gène FGFR3 responsable de l’achondroplasie a été identifié. Une mutation spécifique au niveau de ce gène, nommée G380R, est responsable d'environ 99 % des cas pédiatriques d'ACH. La croissance osseuse est contrôlée par la protéine FGFR3 présente à la surface des cellules du cartilage, les chondrocytes, mais aussi des ostéoblastes, les formes précoces de cellules osseuses. La mutation G380R, ainsi que d'autres mutations similaires, entraînent une activité accrue de FGFR3, ce qui perturbe la chondrogenèse dans la plaque de croissance et altère l'allongement des os longs.
Depuis cette découverte, le laboratoire de bases moléculaires et physiopathologiques des ostéochondrodyslasies à l’Institut Imagine, co-dirigé par les Dr Laurence LEGEAI-MALLET et Pr Valérie CORMIER-DAIRE, explore les mécanismes dérégulés par l’altération de ce gène. Elles mettent au point des modèles cellulaires et animaux pour tester des molécules et développer de nouvelles options thérapeutiques.
Le vosoritide (utilisé en injection sous-cutanée quotidienne), un analogue du peptide C-natriurétique qui agit exclusivement sur la voie des MAP kinases, a été approuvé en 2021 dans le traitement de l'ACH, chez les patients âgés de 2 ans et plus avec des plaques de croissance sont ouvertes. Afin de proposer une thérapie orale ciblant spécifiquement la voie du développement osseux, l'infigratinib, un inhibiteur pan-FGFR1/2/3, a été
étudié dans un modèle de souris imitant l'ACH ( Fgfr3 Y367C/+ ) et est actuellement en essais cliniques.
Parallèlement, le TYRA-300, un inhibiteur oral hautement sélectif de FGFR3, est en étude clinique de phase 1 sous le nom de SURF301 (Study in Untreated and Resistant FGFR3+ Advanced Solid Tumors). Grâce à son profil de spécificité, il pourrait offrir une fenêtre thérapeutique favorable en minimisant les toxicités anticipées des inhibiteurs pan-FGFR.
Pour évaluer le potentiel préclinique du TYRA-300, un modèle murin reproduisant les principales caractéristiques de l'ACH a été utilisé. Ce modèle de souris Fgfr3Y367C/+ se distingue par une petite taille et un déficit de croissance affectant à la fois l'ossification endochondrale et membranaire.
Le traitement avec TYRA-300 a significativement augmenté la longueur corporelle des souris Fgfr3Y367C/+ de 17,9 % par rapport aux témoins, ainsi que la longueur du fémur (+22,6 %), du tibia (+33,0 %) et des vertèbres L4-L6 (+23,5 %). Des améliorations du diamètre du foramen magnum ont également été observées. La coloration histologique a indiqué que le TYRA-300 a restauré l'architecture de la plaque de croissance en améliorant la prolifération et la différenciation des chondrocytes.
La Food and Drug Administration (FDA, États-Unis) a accordé au TYRA-300 la désignation de médicament orphelin dans le traitement de l'ACH. En s'appuyant sur les données de l'étude SURF-301 et des données précliniques supplémentaires, le laboratoire TYRA prévoit d’initier une étude clinique de phase 2 sur l'achondroplasie pédiatrique courant 2024.
Pour en savoir + : Low-dose infigratinib increases bone growth and corrects growth plate abnormalities in an achondroplasia mouse model.
Demuynck B, Flipo J, Kaci N, Dambkowski C, Paull M, Muslimova E, Shah BP, Legeai-Mallet L. J Bone Miner Res. 2024 Apr 9:zjae051. doi: 10.1093/jbmr/zjae051. Lamoine C, et al. ICCBH, 2024
Optimiser les RDV médicaux avec le nouveau guide pratique pour les adultes atteints d'achondroplasie
L’European Achondroplasia Forum (EAF) a élaboré un guide de consultation destiné aux adultes atteints d'achondroplasie, à apporter lors des rendez-vous de routine avec le médecin référent.
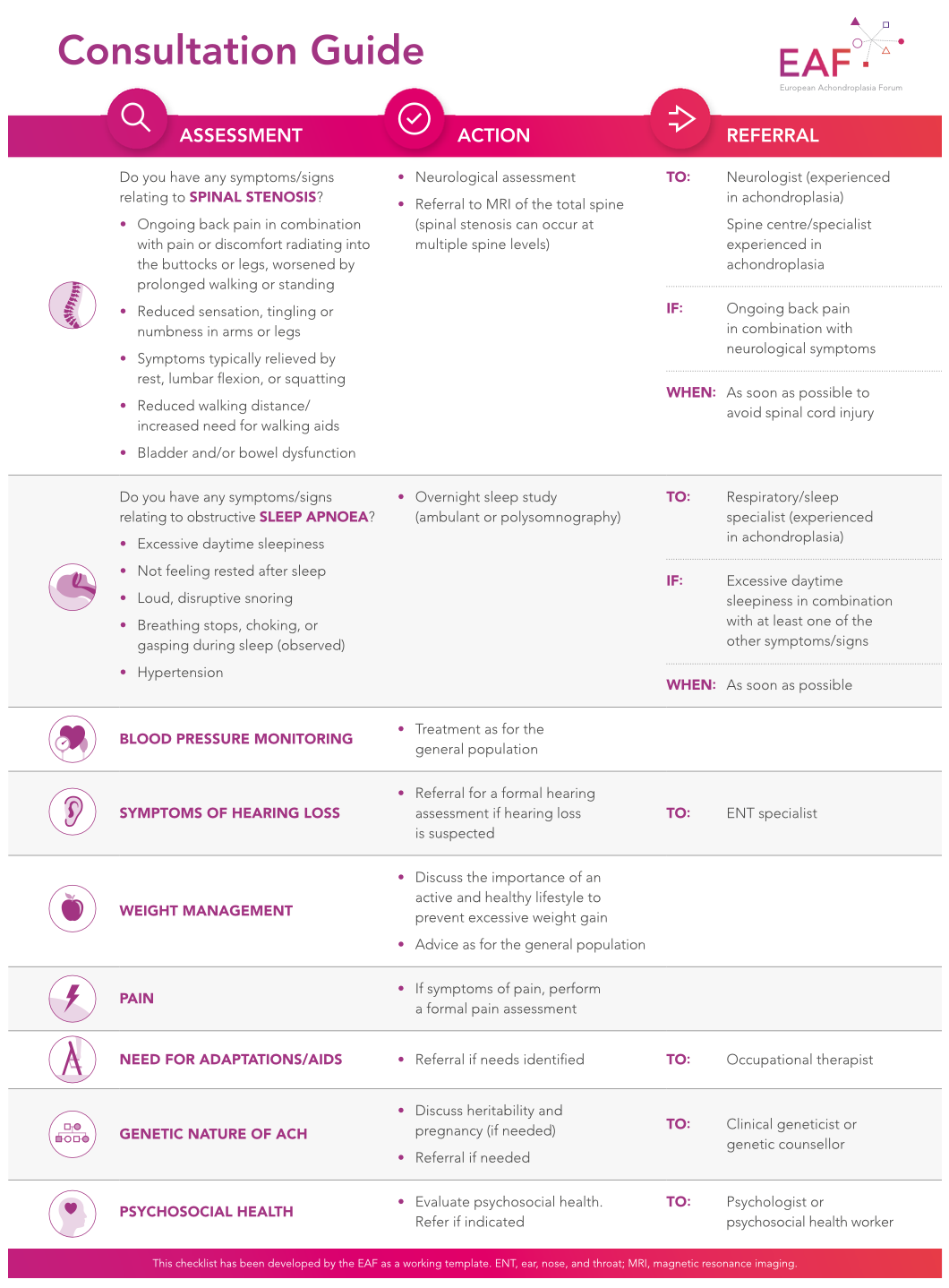
L'achondroplasie est une maladie chronique qui nécessite une prise en charge tout au long de la vie. Il est largement admis que les nourrissons et les enfants atteints d'achondroplasie doivent être suivis par une équipe multidisciplinaire spécialisée. Cependant, de nombreuses personnes perdent le suivi médical lors de la transition des soins pédiatriques vers les soins adultes, et il n'existe pas encore d'approche standardisée pour la prise en charge des adultes, malgré la récente parution de directives internationales de consensus.
Pour répondre à ce besoin, l’European Achondroplasia Forum (EAF) a conçu un guide de consultation destiné aux adultes atteints d'achondroplasie. Conçu pour être utilisé lors des consultations avec le médecin référent, ce guide aide les patients à mieux gérer leur maladie au quotidien. Il souligne les symptômes essentiels à surveiller, tels que la sténose spinale et l'apnée obstructive du sommeil, qui sont à la fois fréquentes et potentiellement graves.
Le guide inclut également des conseils à l'attention des médecins généralistes, indiquant à quel moment il est nécessaire d'orienter les patients vers des spécialistes, afin de faciliter le suivi et le soutien des personnes atteintes. En encourageant une approche proactive dans la gestion de leur santé, ce guide vise à rendre les patients plus autonomes et mieux informés.
Il devrait être remis aux jeunes atteints d'achondroplasie lors de la transition des soins pédiatriques vers les soins pour adultes, dans le but de les aider à comprendre leur maladie, à reconnaître les symptômes clés et les signaux d'alerte.
Pour en savoir + : European Achondroplasia Forum Practical Considerations for Following Adults with Achondroplasia
Fredwall S, AlSayed M, Ben-Omran T, Boero S, Cormier-Daire V, Fauroux B, Guillén-Navarro E, Innig F, Kunkel P, Lampe C, Maghnie M, Mohnike K, Mortier G, Pejin Z, Sessa M, Sousa SB, Irving M.Adv Ther. 2024 Jul;41(7):2545-2558. doi: 10.1007/s12325-024-02880-3.
La greffe de moelle osseuse allogénique : le coup de boost dans la dysplasie cranio-métaphysaire
La recherche concernant la prise en charge de la dysplasie cranio-métaphysaire (DCM) a fait un pas en avant, grâce aux travaux d’une équipe de l’hôpital Necker-Enfants Malades (AP-HP). Cette avancée a été récemment soulignée dans une publication dans le Lancet.
La dysplasie cranio-métaphysaire (DCM) autosomique dominante est une maladie rare qui affecte la résorption osseuse et se caractérise par un épaississement progressif des os crânio-faciaux entraînant des répercussions sur la forme du visage et une déficience visuelle et auditive sévère due à la compression des nerfs crâniens. Jusqu'à présent, la prise en charge médicale se limitait à des mesures symptomatiques, notamment des interventions chirurgicales invasives.
Une découverte récente a révélé l'impact potentiel des greffes de cellules souches hématopoïétiques allogéniques dans le traitement de la DCM autosomique dominante. Traditionnellement, il était difficile d'envisager que cette approche puisse restaurer un phénotype osseux, car les ostéoblastes, responsables de la formation osseuse, ne proviennent pas des cellules souches hématopoïétiques. Cependant, il a été suggéré que la protéine Ank, régulant la formation et la résorption des os, pourrait influencer la fonction des ostéoblastes et des ostéoclastes.
Des études antérieures ont montré que la transplantation expérimentale de moelle osseuse réduisait l'hyperossification chez des souris génétiquement modifiées pour présenter un phénotype de DCM (Ank knockin). Ces résultats ont conduit à l'hypothèse que les greffes de cellules souches hématopoïétiques allogéniques pourraient également être bénéfiques pour les patients humains atteints de DCM autosomique dominante.
Le cas rapporté par l’équipe de l’hôpital Necker-Enfants Malades (AP-HP) concerne une jeune patiente, chez qui une mutation hétérozygote de novo dans le gène ANKH, homologue humain du gène d'ankylose progressive chez la souris, a été identifiée, confirmant le diagnostic de DCM. Les résultats montrent que la greffe de cellules souches hématopoïétiques allogéniques a non seulement atténué l'ostéocondensation (l’épaississement anormal des os), mais a aussi préservé la fonction neurosensorielle, améliorant ainsi la qualité de vie de la patiente.
Cette avancée souligne l'importance d'un diagnostic précoce et d'une prise en charge rapide, avant l’apparition de complications sévères telles que l'hyperostose crânienne et la compression des nerfs crâniens. Des recherches futures pourraient encore améliorer ces résultats et offrir de nouvelles perspectives pour la prise en charge de cette maladie complexe.
Pour en savoir + : Allogeneic bone marrow transplantation in craniometaphyseal dysplasia.
Morelle G, Breton S, Robert MP, Michot C, Boussard C, Cormier-Daire V, Moshous D. Lancet. 2024 May 11;403(10439):1893-1894. doi: 10.1016/S0140-6736(24)00803-1.


